

Description
Keratitis-ichthyosis-deafness (KID) syndrome is characterized by eye problems, skin abnormalities, and hearing loss.
People with KID syndrome usually have keratitis, which is inflammation of the front surface of the eye (the cornea). The keratitis may cause pain, increased sensitivity to light (photophobia), abnormal blood vessel growth over the cornea (neovascularization), and scarring. Over time, affected individuals experience a loss of sharp vision (reduced visual acuity); in severe cases the keratitis can lead to blindness.

Most people with KID syndrome have thick, hard skin on the palms of the hands and soles of the feet (palmoplantar keratoderma). Affected individuals also have thick, reddened patches of skin (erythrokeratoderma) that are dry and scaly (ichthyosis). These dry patches can occur anywhere on the body, although they most commonly affect the neck, groin, and armpits. Breaks in the skin often occur and may lead to infections. In severe cases these infections can be life-threatening, especially in infancy. Approximately 12 percent of people with KID syndrome develop a type of skin cancer called squamous cell carcinoma, which may also affect mucous membranes such as the lining of the mouth.
Partial hair loss is a common feature of KID syndrome, and often affects the eyebrows and eyelashes. Affected individuals may also have small, abnormally formed nails.
Hearing loss in this condition is usually profound, but occasionally is less severe.
Frequency
KID syndrome is a rare disorder. Its prevalence is unknown. Approximately 100 cases have been reported.
Causes
KID syndrome is caused by mutations in the GJB2 gene. This gene provides instructions for making a protein called gap junction beta 2, more commonly known as connexin 26. Connexin 26 is a member of the connexin protein family. Connexin proteins form channels called gap junctions that permit the transport of nutrients, charged atoms (ions), and signaling molecules between neighboring cells that are in contact with each other. Gap junctions made with connexin 26 transport potassium ions and certain small molecules.
Connexin 26 is found in cells throughout the body, including the inner ear and the skin. In the inner ear, channels made from connexin 26 are found in a snail-shaped structure called the cochlea. These channels may help to maintain the proper level of potassium ions required for the conversion of sound waves to electrical nerve impulses. This conversion is essential for normal hearing. In addition, connexin 26 may be involved in the maturation of certain cells in the cochlea. Connexin 26 also plays a role in the growth and maturation of the outermost layer of skin (the epidermis).

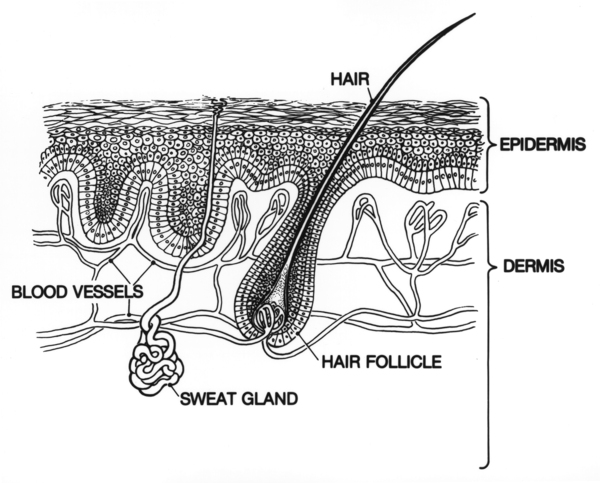
The GJB2 gene mutations that cause KID syndrome change single protein building blocks (amino acids) in connexin 26. The mutations are thought to result in channels that constantly leak ions, which impairs the health of the cells and increases cell death. Death of cells in the skin and the inner ear may underlie the ichthyosis and deafness that occur in KID syndrome. It is unclear how GJB2 gene mutations affect the eye.

Because at least one of the GJB2 gene mutations identified in people with KID syndrome also occurs in hystrix-like ichthyosis with deafness (HID), a disorder with similar features but without keratitis, many researchers categorize KID syndrome and HID as a single disorder, which they call KID/HID. It is not known why some people with this mutation have eye problems while others do not.
Inheritance
KID syndrome is usually inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In some cases, an affected person inherits the mutation from one affected parent. However, most cases result from new mutations in the gene and occur in people with no history of the disorder in their family.


A few families have had a condition resembling KID syndrome with an autosomal recessive pattern of inheritance. In autosomal recessive inheritance, both copies of a gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. Affected individuals in these families have liver disease, which is not a feature of the autosomal dominant form. The autosomal recessive condition is sometimes called Desmons syndrome. It is unknown whether it is also caused by GJB2 gene mutations.

Other Names for This Condition
- Ichthyosiform erythroderma, corneal involvement, and deafness
- Keratitis, ichthyosis, and deafness
- KID syndrome
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Lee JR, White TW. Connexin-26 mutations in deafness and skin disease. Expert Rev Mol Med. 2009 Nov 19;11:e35. doi: 10.1017/S1462399409001276. Citation on PubMed
- Mazereeuw-Hautier J, Bitoun E, Chevrant-Breton J, Man SY, Bodemer C, Prins C, Antille C, Saurat JH, Atherton D, Harper JI, Kelsell DP, Hovnanian A. Keratitis-ichthyosis-deafness syndrome: disease expression and spectrum of connexin 26 (GJB2) mutations in 14 patients. Br J Dermatol. 2007 May;156(5):1015-9. doi: 10.1111/j.1365-2133.2007.07806.x. Epub 2007 Mar 23. Citation on PubMed
- Richard G, Rouan F, Willoughby CE, Brown N, Chung P, Ryynanen M, Jabs EW, Bale SJ, DiGiovanna JJ, Uitto J, Russell L. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am J Hum Genet. 2002 May;70(5):1341-8. doi: 10.1086/339986. Epub 2002 Mar 22. Citation on PubMed or Free article on PubMed Central
- van Steensel MA. Gap junction diseases of the skin. Am J Med Genet C Semin Med Genet. 2004 Nov 15;131C(1):12-9. doi: 10.1002/ajmg.c.30030. Erratum In: Am J Med Genet C Semin Med Genet. 2006 Feb 15;142(1):58. Citation on PubMed
- Xu J, Nicholson BJ. The role of connexins in ear and skin physiology - functional insights from disease-associated mutations. Biochim Biophys Acta. 2013 Jan;1828(1):167-78. doi: 10.1016/j.bbamem.2012.06.024. Epub 2012 Jul 13. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.